In Ornstein and Zernike’s seminal work, Accidental deviations of density and opalescence at the critical point of a single substance, in: KNAW, Proceedings, 17 II, 1914, pp. 793-806, it was shown that the total correlation between molecules in a statistical mechanical system must obey an integral equation of the form,
where is the total correlation function between particles and (note that , where is the more familiar radial distribution function) and is the ‘direct’ correlation between particles and . In this article, I will try and demystify this equation using physical arguments from Ornstein and Zernike’s original paper as well as the more recent formulations described in Liquid State Theory from Hansen and McDonald as well as Santos’ A Concise Course on the Theory of Classical Liquids.
Ornstein and Zernike’s Original Derivation
The motivation for Ornstein and Zernike’s work was from an original paper on critical behavior of statistical mechanical systems from Smoluchowski, with the main problem being that his volume elements, of which he used to partition up a system of particles, were assumed to be mutually independent of one another. At this stage, the whole premise might already sound a little fishy. We know that liquids interact with each other strongly, so of course this mutual independence assumption must be a problem. Why are we even talking about volume elements in the first place? And what is meant by their mutual independence?
Ornstein and Zernike implore us to consider a statistical mechanical system composed of a finite set of volume elements containing a finite number of particles so that the total volume is,
and the total number of particles is,
Nice! Intuitively, all we have done is taken a system of particles and discretized the position space into finite volume elements which contain a finite set of particles. Of course, in an atomic systems the particles are always moving, and indeed, always passing from one volume element to another. In general, we will say that the mean square deviation of the number of particles in a given volume element based on the atomic motion is, . If the number of particles in the element of volume is large, then this mean square deviation is approximately equal to the variance of the number of particles in the volume element. If we consider that our atomic system is a subsystem of some larger collection of particles, then we can express the mean number of particles in our system as the sum of the mean number of particles in each volume element so that,
and therefore the mean square deviation of the number of particles in our system is,
which has a complicated expansion given by,
and, assuming that the cross correlations are not related and that the number of particles are equal across all of the volume elements (in which we will say there are in total) gives,
The biggest problem here is the cross terms which appear in our expansion are at odds with a mutual independence approximation. Indeed, if the system were truly independent, then there would be no contribution to the mean square deviation from pairs of different volume elements. This forms the key difference between Ornstein and Zernike’s formulation and that of Smulochowski.
The Ornstein-Zernike Equation
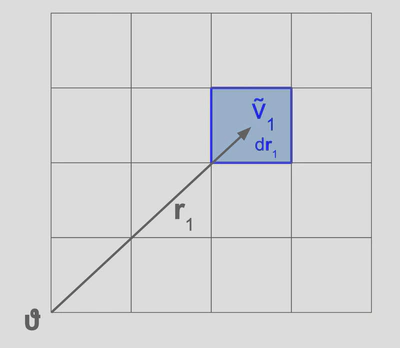
Now, consider that we take our system and subdivide into an infinite number of volume elements and select one to be our reference, . We want to ask how fluctuations in our local volume element influence the densities of all other volume elements in the system, which is of course related to how much the particles in our reference volume interact with all the other particles. On average, we claim that the average density in our volume element, , is linearly related to the density in all other volume elements so that,
where are coefficients of the linear equation relating the local density to that of the other volume elements. Generally, we could write this expression as a Taylor expansion over terms of increasing order, although Ornstein and Zernike claim that the linear term is sufficient for their purposes. Although we will table this for now, it is an interesting question as to whether such an expansion is truly sufficient for statistical mechanics of liquids and whether or not this causes issues with the OZ relation methods. Moving on, we assume that this function is continuous and well-behaved over the so-called ‘sphere of attraction’ of the system, so we can rewrite this sum as an integral of continuous functions such that,
where the integral is understood to go over all of . Note that the ‘sphere of attraction’ simply refers to the range of direct influence of the particles of on the other particles in the system. We would expect such a sphere to be finite, since the direct influence of particles in our reference volume is an interatomic potential that is at its strongest Coulombic with dependence. However, note that this assumption leads to difficulties with the Fourier transform (see section ). More generally however, the function is not actually continuous if we consider that the particles are point particles, the reason being that if we create an infinite number of volume elements for the system that there will be some volume elements with particles and others with none. Such a problem is avoided by rewriting the expressions in terms of a new function which will be introduced later. However, in Ornstein and Zernike’s original work, there is not a satisfactory discussion of why such a function should be continuous at all, which would require further nuance and discussion found in the free energy functional approach discussed later.
Let us now suppose that the density in our volume element is known to be and write the average density at any other point in space as,
the function being dependent on, of course, the position in the system and choice of reference. What then is given that we know ? Selecting a known value of at would give us a of,
which is just the definition of selected at a point shifted in space from our original reference. The average value at is then simply,
Applying the original integral with an assumed known is then,
where the integral contribution at the selected particle position is written explicitly outside of the integral (known from the linear approximation to the Taylor expansion above) since is not defined there. Finally, note that this must be true for any choice of and that is linear in , meaning that we can pull out a factor of for both instances of so that,
In modern symbols, the is actually just the total correlation between densities in volume elements of the system, which we write as , and represents the direct coupling between volume elements, which we write as . In modern symbols, the Ornstein-Zernike equation emerges,
Physical Interpretation
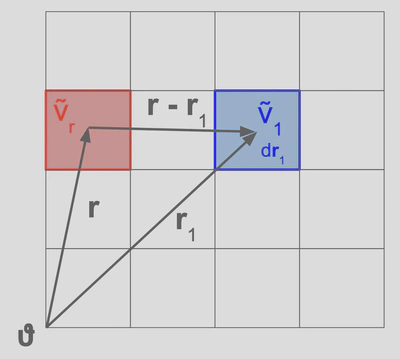
What exactly does this equation mean? Starting with the total correlation function , we notice that this function is simply the average particle density of the system at position within a spherical coordinate system,
According to the Ornstein-Zernike equation, this total correlation function is fully described by three functions: the direct correlation function , the direct correlation function , and the total correlation function . These functions only make sense when we consider that the integral will go over all of the other volume elements of the system, specifically for all . Our new picture is then,
Now let’s consider each of the terms. The direct correlation function is a function of linear coupling constants which relate the particle density at the origin to all other volume elements in the system. There are no cross terms in the expansion, so this function only keeps track of the ‘direct’ coupling between volume elements, hence the name. is therefore the direct coupling between the reference origin and some other volume element. The other direct correlation function has the same interpretation, but now it represents the direct coupling between the reference and a different volume element not centered at . Finally, the total correlation is just the density of a volume element from the reference volume element between the difference in the vectors .
Finite Leads to Nonphysical Fourier Transforms
There are some problems with the mathematical development presented by Ornstein and Zernike. In a short phone call with Harry W. Sullivan, who just traveled to University of Minnesota to start a doctoral program, it was noted that the assumption of a cut-off in may actually be at odds with Fourier representations of the total correlation function. To see this, consider that the modified Henkel transform (or spherical Bessel transform) of the real-space total correlation function is directly related to the total structure factor, a quantity which is observable experimentally via scattering experiments. Thus, assuming that the total correlation function is isotropic, we can write the modified Henkel transform of the total correlation function as,
where is the Bessel function of the first kind of order . If there is a point at which the total correlation abruptly becomes zero at some , then, expanding in a Fourier Bessel series gives,
where are constants and is the root of the Bessel function. The sum and integral can be exchanged since they are linear, giving the following,
which has as a solution a delta-train,
This result, of course, is at odds with experimental measurement of the total structure factor and with physical intuition of the problem. Hence, the abrupt sending of the function to zero is not a mathematically reasonable assumption to describe the theory of liquids.
Conclusions from Ornstein and Zernike’s Original Work
In summary, Ornstein and Zernike’s original paper is a work of genius that resulted in an integral relation that has remained relevant to liquid state physics for over a century. Indeed, reading this manuscript one gets the sense that the authors did not understand how monumental this work would become in the development of modern physics. However, the mathematical development in their manuscript is not rigorous and therefore encounters some potential pitfalls, but nonetheless was later verified to be correct anyway without the shaky assumptions that derail the extensibility of their method.
The Gibbs Interpretation
One of the great issues with Ornstein and Zernike’s original derivation is that the direct correlation function is constructed with non-rigorous assumptions and doesn’t have a physical interpretation. However, these deficiencies can be addressed with the statistical mechanics methods of Gibbs. First, recall that the phase space probability density, , is a function so that is the probability at time that the system will be in the -dimensional phase space element . By defining a specific physical system, we can ascribe a functional form to this probability distribution. For instance, if we take a collection of particles at equilibrium with a heat bath of fixed temperature but that cannot exchange particles, we obtain the canonical probability distribution,
where is the familiar -particle canonical partition function and the subscript of 0 for indicates that the system is at equilibrium and therefore does not depend on time.
Generally, the -particle phase space probability density function contains unnecessary information that can be integrated out to obtain so-called reduced phase space distribution functions in the following way,
which has an obvious physical interpretation: gives the probability of finding the system in a reduced phase space element irrespective of all other particle positions and momenta. Now, in general this reduced phase space probability density at equilibrium is separable into kinetic and potential parts () so that,
where the is the equilibrium -particle density function so that is the probability of finding a set of particles in reduced phase space volume element irrespective of the positions of all other particles and irrespective of all momenta.
Direct Correlations as Derivatives of Free Energy
The Gibbs interpretation offers us more mathematical rigor and precision that resolves many of the issues we have seen in Ornstein and Zernike’s original manuscript. First, we will discuss the physical interpretation of the total correlation function in the Gibbs interpretation. Intuitively, the total correlation function, , which appears in the OZ equation, describes the ensemble average distribution function of any two particle densities in the system. However, this quantity is defined in terms of the equilibrium -particle density function in the following way,
which, for a reduced 2-particle density function, can be written as,
and finally as our function,
Notice that this function is defined in terms of a 2-particle density function, which contains contributions from both individual particle densities and the intermediate particles as in our original picture from the Ornstein-Zernike manuscript. Notice we have completely avoided the difficulties of assuming that the density function is continuous (when in fact, it is not), since is equally described by an ensemble average that is continuous for liquids at equilibrium.
So what of Ornstein and Zernike’s coupling constant function, , under the Gibbs interpretation? Perhaps surprisingly, is exactly equal to a second partial functional derivative of the excess free energy functional, (recall that the excess part of the intrinsic free energy is just the contribution to the free energy due to particle interactions), so that,
Such a physical interpretation of the direct correlation is extremely satisfying for the following reasons. First, we see that indeed the direct correlation does not involve reduced particle density distributions higher than order 1, meaning that there is no contribution from intermediate particles (we had the same situation in Ornstein and Zernike’s picture previously). However, now can be thought of as a response of the excess intrinisic free energy to changes in the single particle density distributions, rather than just the linear coupling terms of a truncated Taylor expansion. Such a construction does not require to abruptly vanish, resolving the issue with the modified Henkel transform. Finally, we now see how could even be used to approximate the intrinsic free energy functional and hence describe entirely the thermodynamics of a model fluid.
The OZ Equation in the Gibbs Interpretation
See Section 3.5 of Theory of Simple Liquids by Hansen and McDonald.
Numerical Implementations
The Isotropic OZ Relation and the Bridge Function
In practice, we usually consider liquids to be isotropic, allowing us to reduce the vector quantities introduced earlier into just the distance between particles, , to give an isotropic Ornstein-Zernike relation,
which you may notice is just a convolution of with ,
which under Fourier transform is simply,
or rearranging,
Finally, diagrammatic techniques (see \textit{A Concise Course on the Theory of Classical Liquids} by Santos and Section ) and the introduction of the indirect correlation function (), allow us to write the radial distribution as,
where is the so-called, and unkown to us, bridge function arising from elementary diagrams in the integral expansion of . Using the definition of the indirect correlation, we find that,
Noting that , we can subtract the direct correlation from both sides of the equation to obtain,
and using the definition of the indirect correlation and the diagram expansion of ,
which, upon rearranging, gives,
Here a closure relation is introduced to write in terms of the indirect correlation function. Specific examples will be provided in section .
OZ Fourier Transform in Terms of the Indirect Correlation
In terms of the indirect correlation, the OZ equation becomes,
which, upon taking the Fourier transform, gives,
and rearranging,
Iterative Numerical Solvers
We now have everything we need to solve for the pair correlation function , given a potential. The steps we use are:
Guess the indirect correlation function .
Compute the direct correlation function, , using a suitable closure relation for the bridge function.
Fourier transform .
Compute Fourier transform of the indirect correlation, .
Fourier transform to get a new guess for .
Repeat until convergence.
Now, what if we have the total correlation function from experiment but do not know the potential? In this case, we change the algorithm as follows:
Guess the pair potential .
Compute the direct correlation function, , using a suitable closure relation for the bridge function.
Fourier transform .
Compute Fourier transform of the total correlation, .
Fourier transform to get .
Perform iterative refinement scheme to update potential to .
Repeat until the computed matches the experimental data.